Research interests

Research in MASCL integrates synthetic and physical organic chemistry with materials and biological science. The main interest is the development of innovative macrocyclic and supramolecular chemistry. In particular, we focus on the design and synthesis of novel and functional macrocycles which are capable of serving as privileged platform molecules useful for supramolecular chemistry and nanotechnology. Using a multi-faceted approach, we also study non-covalent anion-p interactions as a fundamental motif and exploit them for a driving force to construct a diversity of supramolecular systems. In addition, we strive to develop chiral supramolecular system of inherently chiral macrocyclic compounds. To achieve this goal, we are exploring methods to synthesize and assemble enantiopure inherently chiral macrocyclic compounds. Furthermore, we take advantage of rationally well-designed synthetic macrocycles to explore the formation, reactivity and mechanism of high valent arylcopper compounds, promoting organocopper chemistry and copper catalysis. Finally, we are interested in developing enantioselective biocatalytic and chemical catalytic reactions which allow the efficient and rapid access to chiral molecules of significance in materials and life science.
- 1. Macrocyclic Chemistry of Novel and Functional Macrocyclic Molecules
- 2. Anion-π Non-Covalent Bond Interactions
- 3. Supramolecular Chemistry of Inherently Chiral Macrocycles
- 4. High Valent Organocopper Chemistry
- 5. Enantioselective Biotransformations of Nitriles
- 6. Tertiary Enamides as Versatile Synthons in Organic Synthesis
1. Macrocyclic Chemistry of Novel and Functional Macrocyclic Molecules
1.1 Macrocyclic Chemistry of Heteracalixaromatics and Coronarenes
Supramolecular chemistry was established through the pioneering works of Pederson, Lehn and Cram on crown ethers, cryptands and spherands. Since then, the design and construction of novel and functional macrocyclic molecules have always been one of the central focuses of study in supramolecular science. Tailor-made synthetic macrocycles offer not only the excellent model systems to study the nature of various non-covalent interactions but also the essential building units for the fabrication of sophisticated (supra)molecular structures, advanced materials and machinery systems. Furthermore, two- and three-dimensional macrocycles with well-defined cavities are unique molecular tools in highly selective synthesis and in the study of reaction mechanisms. Despite numerous synthetic macrocyclic compounds reported in literature, there is a handful of versatile and useful macrocyclic hosts.
We started our studies of macrocyclic and supramolecular chemistry at the beginning of the 21st century with the focus of the development of novel macrocyclic hosts. Taking advantage of nitrogen and oxygen atoms, unlike the methylene groups that are key bridging elements in conventional calixarenes, can adopt different electronic configurations and can form different conjugation with their adjacent aromatic rings, we have devised in sequence two sets of functional macrocyclic hosts, namely the heteracalixaromatics and the coronarenes (Figure 1). In general, heteracalixaromatics consist of meta-arylenes linked with heteroatoms while coronarenes are composed of para-arylenes and heteroatoms in an alternative fashion. These macrocycles support a variety of conformations and provide cavity structures that could not be previously accessed. It is also allowed for rational control over the electronic features of these new macrocyclic hosts.
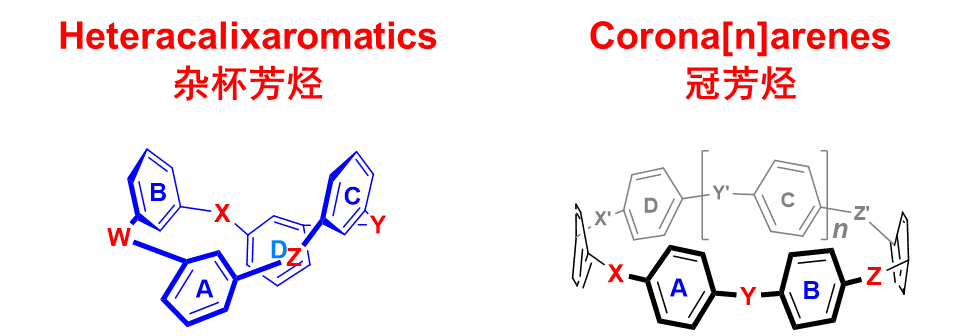
In the past years, we established a “fragment coupling” synthetic strategy that allowed preparation of heteracalixaromatics and coronarenes containing different heteroatom bridges and various aromatic subunits. We also developed a post-macrocyclization protocol, macrocycle-to-macrocycle transformations and functional group transformations that allowed functional groups to be installed onto the macrocyclic skeleton in a regioselective manner. The methods enabled generation of a myriad of tailor-made functionalized heteracalixaromatics and coronarenes. We have demonstrated that heteroatom-linked calix[4](het)arenes adopt the 1,3-alternate conformation while coronarenes give various coronary conformations. Remarkably, heteracalix[4]aromatics and coronarenes would self-tune their cavity sizes to accommodate guest molecules owing to the interplay between the bridging heteroatoms and the constituent aromatic rings.
It is noteworthy that heteracalixaromatics and coronarenes are emerging as privileged macrocyclic hosts, finding use across a wide range of applications from molecular recognition of organic molecules and charged guests to the fabrication of functional assemblies. It has been shown in our laboratory that they able to selectively complex transition metal ions, anions, fullerenes C60 and C70, aliphatic and aromatic alcohols and diols, to stabilize reactive polynuclear silver carbide clusters by encapsulation, and to bind organic ammonium salts forming pseudorotaxane complexes. We have also discovered that azacalix[1]arene[3]pyridines react with copper(II) salts to form structurally well-defined high valent arylcopper compounds. Using these high valent arylcopper compounds as probes, the mechanisms of copper-catalyzed arene C-H bond activation and transformations have been elucidated. The acquired macrocycles are invaluable building blocks that permit fabrication of sophisticated (surpa)molecular architectures including cages, barrels, and liquid crystals. Many other research groups across the world have used heteracalixaromatics and coronarenes to construct MOFs, COFs, LC, photo-responsive materials, spontaneous and selective CO2 sorption porous materials, solid phase extraction materials and HPLC stationary phase. Fabrications of AIE systems, artificial anion channels and transporters, self-assembled stimuli-responsive vesicles, organocatalysts and molecular machines have also been reported.
References
[1] Acc. Chem. Res. 2012, 45, 182-195.
[2] Calixarenes and Beyond, Eds. P. Neri, J. L. Sessler, M.-X. Wang, Springer, 2016.
[3] Sci. China Chem. 2018, 61, 993-1003.
1.2 Zigzag Hydrocarbon Nanobelts and Heteroatom-Embedded Analogs
Zigzag hydrocarbon nanobelts have been fascinating chemists and materials scientists for decades because of their aesthetically appealing molecular structures, outstanding physical properties and intriguing chemical reactivities predicted by theoretical calculations, and potential applications as unique macrocyclic hosts in supramolecular chemistry. They may also serve as templates or seeds to grow structurally well-defined uniform zigzag carbon nanotubes. While there have been continuous computational studies on the structures and properties of belt[n]arenes or [n]cyclacenes since they were proposed as hypothetical molecules in 1954, the synthesis of (partially) conjugated zigzag hydrocarbon belts remains a great challenge with no progress being reported in the past 20 years. As a consequence, the study of these exquisite and extraordinary molecules has been dormant for more than two decades.
It is important to note that, prior to our study, there were various terms to name zigzag hydrocarbon belt molecules. To unify the nomenclatures and also to follow the tradition in naming macrocyclic compounds in supramolecular chemistry, we recommend to use belt[n]arenes to name double-stranded molecules in which aromatic rings are ortho-fused in a linear fashion. Collar[n]arenes, belt[n]enes and belt[n]anes can be named therefore as partially or fully hydrogenated or saturated belt[n]arene derivatives. Moreover, this naming system can be easily extended to heteroaromatic ring-containing belt[n]arene analogs. For example, belt[4]arene[4]pyridine or tetraza-embedded belt[8]arene is a zigzag molecular belt which contains four benzene and four pyridine units.
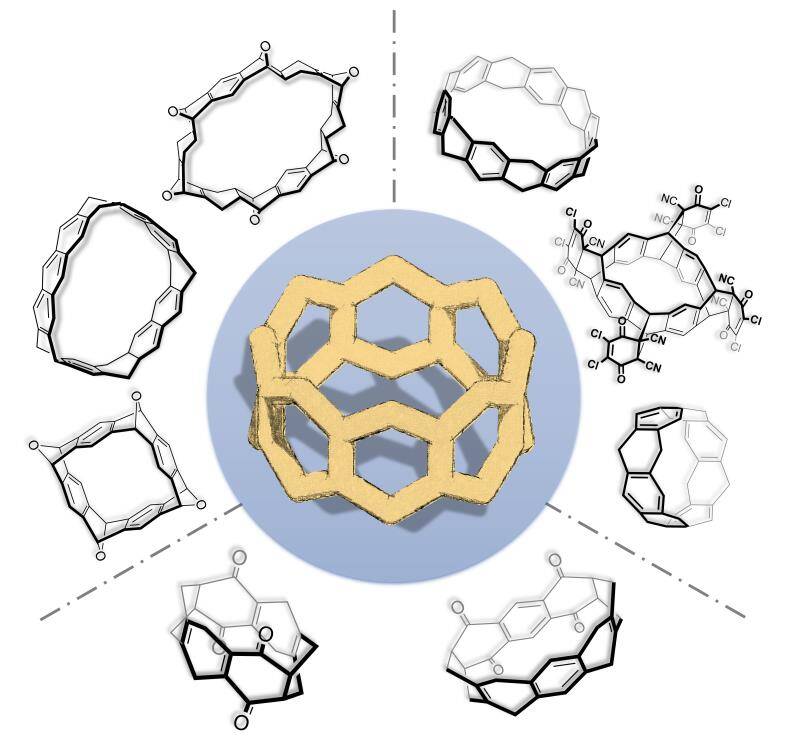
Since we started this MASCL at Tsinghua in 2009, we have been focusing on the synthesis of zigzag hydrocarbon nanobelts and their heteroatom-embedded analogs. We have established successfully a “fjord-stitching” strategy to construct partly hydrogenated belt[n]arenes and their heteroatom-doped analogs through different multiple intramolecular cyclization reactions by taking advantage of resorcin[n]arenes, which are readily available from the condensation between resorcinol and aldehydes, and have a pre-organized cone conformation with pre-installed hydroxy groups. The molecules adopt unique nanobelt structures which are highly strained. Oxidative aromatization of octahydrobelt[8]arene with DDQ led to a belt[8]arene-DDQ4 adduct which underwent most probably retro-Diels-Alder reaction to form the first fully conjugated beltarene under laser irradiation. These works represent a true breakthrough in the design and synthesis of zigzag hydrocarbon nanobelts.
References
[1] CCS Chem. 2020, 2, 916-931.
[2] Nat. Chem. 2021, 13, 402-410.
[3] Chem 2020, 6, 826-929.
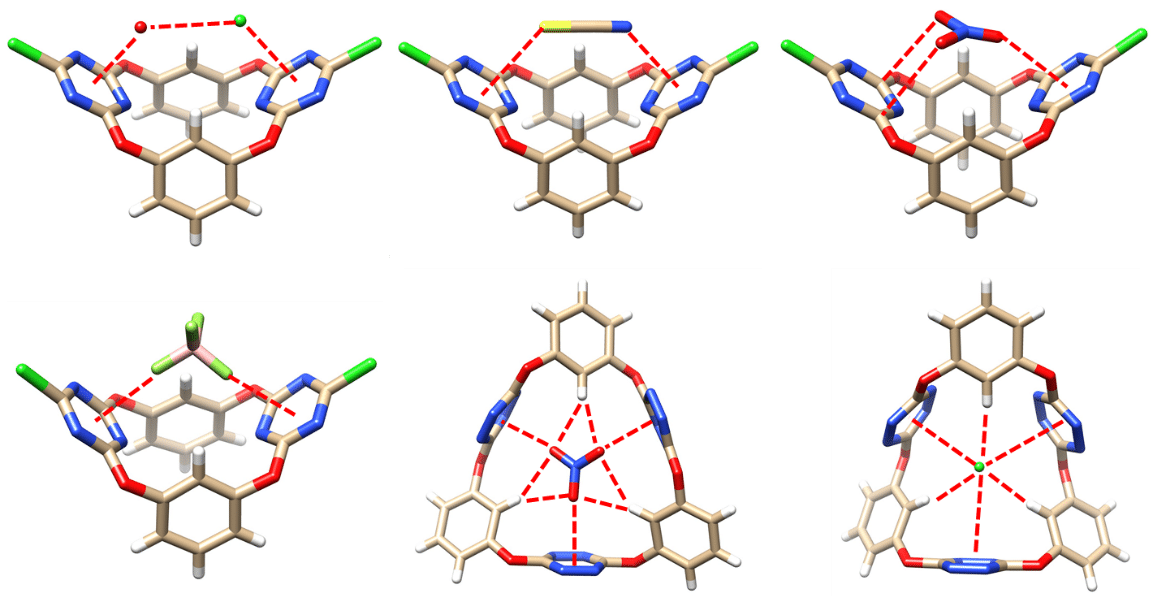
2. Anion-π Non-Covalent Bond Interactions
Noncovalent bonding interactions underlie most supramolecular processes. While hydrogen bonding and π–π interactions are arguably textbook concepts, noncovalent anion−π interactions have only emerged over the last two decades. Since π systems are traditionally considered to be electron rich, the interactions between anions and π systems might be thought to be repulsive. This stands in stark contrast to the acknowledged favorable nature of cation−π interactions. Although a plethora of calculations had been published, the lack of experimental evidence cast doubt on the existence of anion−π interactions between anions and charge-neutral aromatic systems. Utilizing heteracalixaromatics and coronarenes as hosts, we have shown that under certain circumstances anion-π interactions can be attractive and can be used as the basis for supramolecular chemistry effects. We showed in 2008 the first experimental demonstration of an attractive non-covalent bonding interaction between anions and an electron-neutral π system. Later the generality, strength and ubiquity of anion-π interactions have been reported in 2013. Recently, we have revealed the cooperative effect of multiple anion-π interactions, and an interdependent and synergistic effect between anion-π and unconventional hydrogen bond interactions in anion recognition. The importance and utility of anion-π interactions is now widely recognized, both as a fundamental motif likely exploited by nature, but also a driving force that may be used to construct a diversity of supramolecular systems.
References
[1] Angew. Chem. Int. Ed. 2008, 47, 7485-7488.
[2] J. Am. Chem. Soc. 2013, 135, 892-897.
[3] Angew. Chem. Int. Ed. 2018, 57, 6536-6540.
[4] Angew. Chem. Int. Ed. 2020, 59, 8078-8083.
[5] Angew. Chem. Int. Ed. 2020, 59, 23716-23723.
[6] Acc. Chem. Res. 2020, 53, 1364-1380.
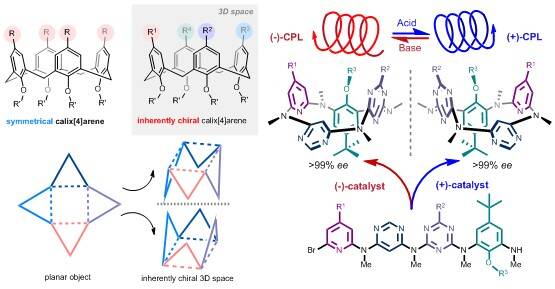
3. Supramolecular Chemistry of Inherently Chiral Macrocycles
Molecules are defined as chiral ones when they do not have element of symmetry such as the plane (s), center (I) and alternating axis (Sn) of symmetry. In general, most of the conventional chiral molecules are classified into central, axial, planar chiral molecules, and helical molecules. Böhmer suggested in 1994 “inherently chiral calixarenes” to describe chiral calixarenes devoid of a plane of symmetry or an inversion center in a molecule. When calix[4]arenes are composed of four different aromatic rings, or to introduce different substituents on either the lower rim or upper rim positions, inherently chiral calix[4]arenes are generated due to the loss of molecular symmetry [s, I, Sn]. Actually, introduction of a curvature in an ideal planar structure that is devoid of symmetry in its bidimensional representation will result in inherent chirality. By contrast to conventional chiral macrocyclic molecules which are composed of chiral subunits, inherently chiral macrocycles provide asymmetric three-dimensional structures and form a diversity of chiral spaces which would be unique and invaluable in the study of chemistry, materials and life science. We have been developing the supramolecular chemistry of inherently chiral macrocycles for years with the focuses on the catalytic asymmetric syntheses, structures and the functions of novel macrocyclic compounds. We have reported, for instance, a strategy to construct inherently chiral macrocycles, namely, ABCD-type heteracalix[4]aromatics, through a catalytic enantioselective intramolecular C-N bond forming reaction. The method enables the preparation of highly enantiopure compounds which were only available in an analytic amount via resolution on the chiral stationary phase of a column using HPLC. While the versatile applications await study, the resulting ABCD-type heteracalix[4]aromatics have been shown to display excellent and pH-triggered switchable electronic circular dichroism and circularly polarized luminescence properties. We believe that with the advent of new and efficient synthetic methods, the chemistry of inherently chiral macrocycles will flourish in the years to come.
Reference
[1] J. Am. Chem. Soc. 2020, 142, 14432-14436.
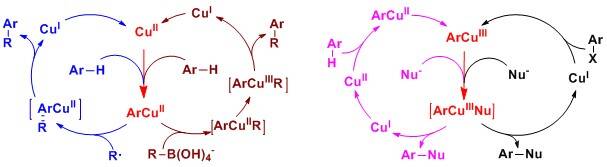
4. High Valent Organocopper Chemistry
Copper salts are naturally abundant, inexpensive and less-toxic in comparison to precious metals. As the surrogate of noble metal catalysts, copper salts have been shown to exhibit remarkable versatility in catalyzing and promoting organic reactions. Despite significant developments in synthetic methodology, the mechanism for copper catalysis remains elusive. Hypothetic reaction pathways such as two electron Cu(III)/Cu(I) and Cu(II)/Cu(0) catalytic cycles, and the Cu(II)/Cu(I) catalytic cycle through a single-electron transfer process have been invoked to diagram the catalytic C-H bond transformations because of the formidable challenges to isolate and characterize reactive or transient high valent organocopper intermediates. In fact, organocopper chemistry has long been dominated by organocopper(I) compounds. Our journey to the chemistry of high valent organocopper compounds started with a serendipitous discovery of the facile formation of a stable arylcopper(III) compound when we examined routinely the titration of tetraazacalix[1]arene[3]pyridine with Cu(ClO4).6H2O. Our curiosity about the reaction pathways for direct arene C-H bond cupration, the intrinsic reactivities of high valent organocopper species, and mechanistic aspects of copper-promoted C-H bond activation and functionalization prompted us to initiate a systematic study on novel organocopper chemistry. We have explored the reactivities of both arylcopper(II) and arylcopper(III) compounds, demonstrating their versatility and uniqueness in chemical synthesis. Novel and fascinating arene C-H transformations under copper catalysis have been developed. Using acquired high valent arylcopper compounds as molecular probes, and employing the functionalizations of tetraazacalix[1]arene[3]pyridines as model reactions, we have been able to reveal the diverse mechanisms of copper-catalyzed or -mediated arene C-H bond reactions. Elusive reaction pathways of some copper-promoted C-X bond activations have also been unraveled. Without doubt, studies of the synthesis, reactivity, and catalysis of high valent organocopper compounds have been reshaping the field of organocopper chemistry.
Reference
[1] Chem. Commun. 2009, 2899-2901.
[2] J. Am. Chem. Soc. 2014, 136, 6326-6332.
[3] J. Org. Chem. 2016, 81, 10404-10410.
[4] J. Am. Chem. Soc. 2018, 140, 5579-5587.
[5] J. Am. Chem. Soc. 2019, 141, 18341-18348.
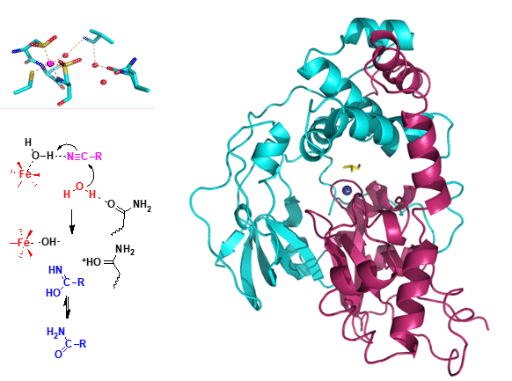
5. Enantioselective Biotransformations of Nitriles
Hydration and hydrolysis of nitriles are valuable synthetic methods used to prepare carboxamides and carboxylic acids. In nature, biological systems degrade nitriles via two distinct pathways: The nitrilases catalyze direct hydrolysis of nitriles to afford carboxylic acids with release of ammonia, and the nitrile hydratases catalyze the conversion of nitriles into carboxamides which furnish carboxylic acids via hydrolysis in the presence of the amidases. Remarkably, the biological transformations of nitriles are highly efficient, chemo-selective, and environmentally benign. They are in stark contrast to chemical processes which involve harsh reaction conditions, have low selectivity, and generate large amounts of waste. Since the late 1990s, we have conducted an in-depth study of enantioselective biotransformations of nitriles and amides using microbial whole cells as biocatalysts. We have established green, unique and powerful biocatalytic methods for the synthesis of a diversity of carboxylic acids and their derivatives with high enantiopurity. Most of the enantiopure products are not readily available by other synthetic methods. By means of X-ray crystallography, we have elucidated the mechanism of nitrile hydratase. Our study has also led to a reaction model to predict biocatalytic efficiency and enantioselectivity of three- and four-membered ring substrates. Combined with chemical transformations, natural products such as Clausena alkaloids and bioactive heterocyclic compounds have been synthesized efficiently.
Reference
[1] Acc. Chem. Res. 2015, 48, 602-611.
6. Tertiary Enamides as Versatile Synthons in Organic Synthesis
While enamines always enjoy their popularity among synthetic chemists, tertiary enamides had long been ignored and thought of as stable and marginally valuable enamine variants because of the presence of an N-electron-withdrawing acyl group. After analysis of the cross-conjugation system of the molecules, we envisioned that enabling the regulation of the cross-conjugation system by either electronic and steric effects of the substituents on the enamide segment would revive the nucleophilic reactivity of tertiary enamides. Moreover, the initial nucleophilic reaction of tertiary enamides would generate the highly reactive N-acyl iminium intermediates which can either undergo deprotonation or addition reaction with a nucleophile. As a result, the rational design of the tertiary enamides and the control of reaction conditions would result in the selective synthesis of functional organic compounds. Since 2007, we have fully explored various reaction pathways of tertiary enamides focusing on the catalytic asymmetric, multicomponent and tandem synthesis of N-heterocyclic compounds. To our delight, our endeavors have turned the tertiary enamides into a type of versatile synthons in organic synthesis. For example, we have shown that, in the presence of (chiral) catalysts, tertiary enamides exhibit good enaminic reactivity towards functional groups such as epoxides, carbonyls, imines, nitriliums and activated alkynes. Tertiary enamides are also able to undergo catalytic asymmetric difunctionalizations in multicomponent and cascade reaction manners. A wide variety of N-heterocyclic compounds including alkaloids of biological and pharmaceutical relevance have been synthesized successfully. We can foresee that, owing to their easy availability and the excellent designability of the reaction pathways under (asymmetric) catalysis, “stable” tertiary enamides as unique synthons will find extensive applications in the arena of organic synthesis.

Reference
[1] Chem. Commun. 2015, 51, 6039-6049.
[2] Synlett. 2021, early access, DOI: 10.1055/a-1352-6358.